Project 11
- Project Topic Subtitle: Thermal switching and operation of hybrid DNA-origami plasmonic structures and nanopores
- ESR Student Name: Aleksei Overchenko
- State of the art: state-of-the-art-2022
- Selected Outputs / References:
Solid-state and DNA-origami hybrid nanopores
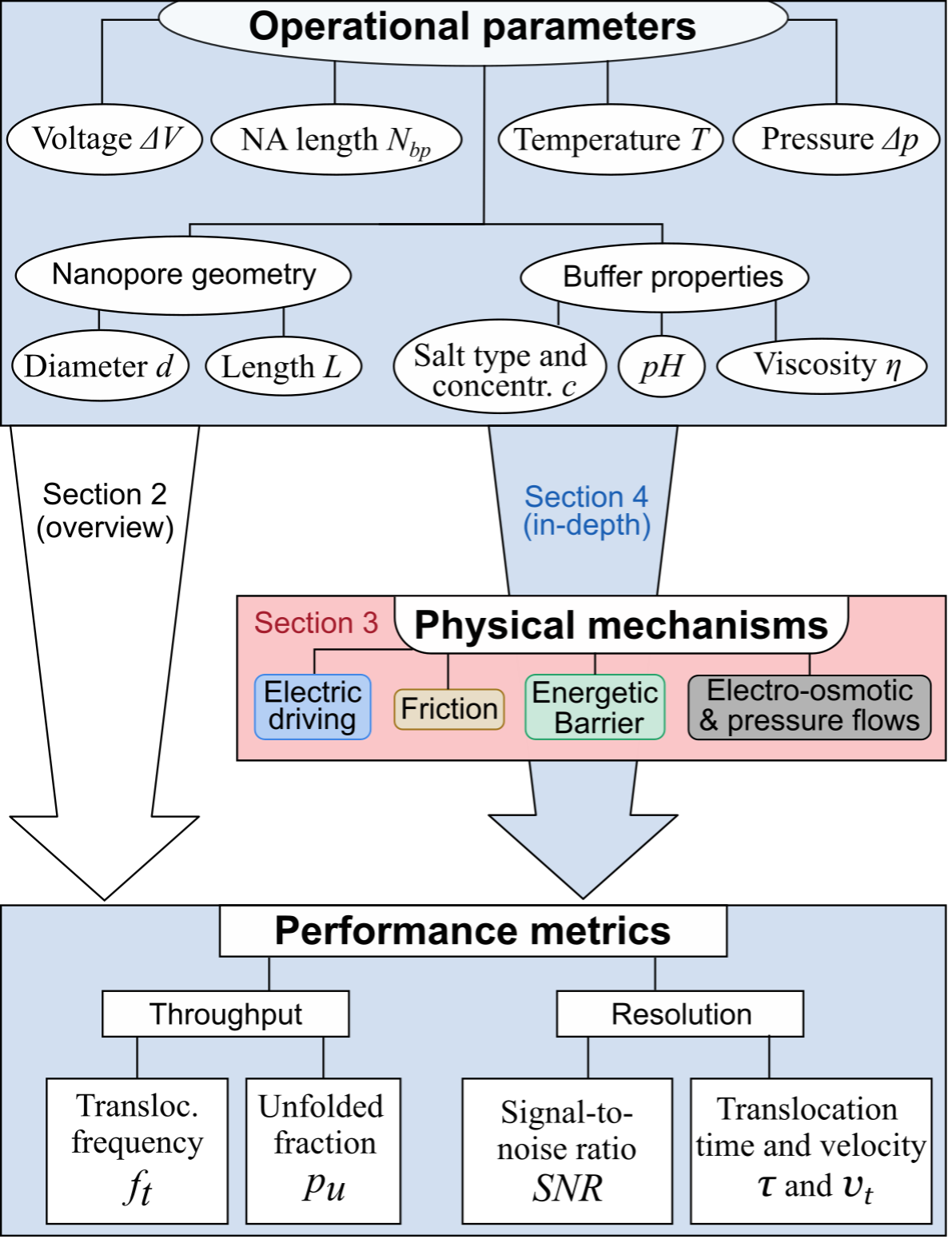
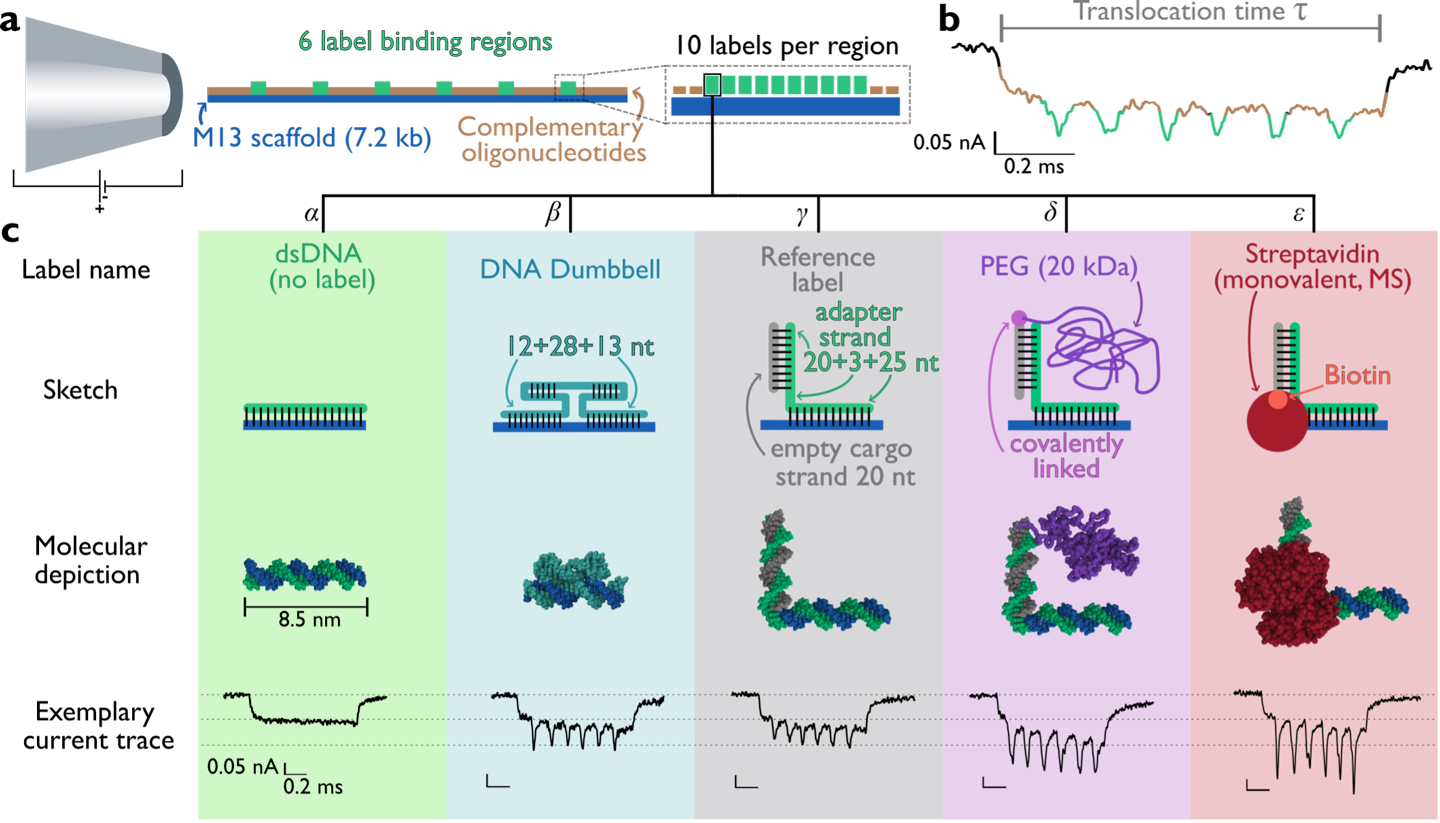
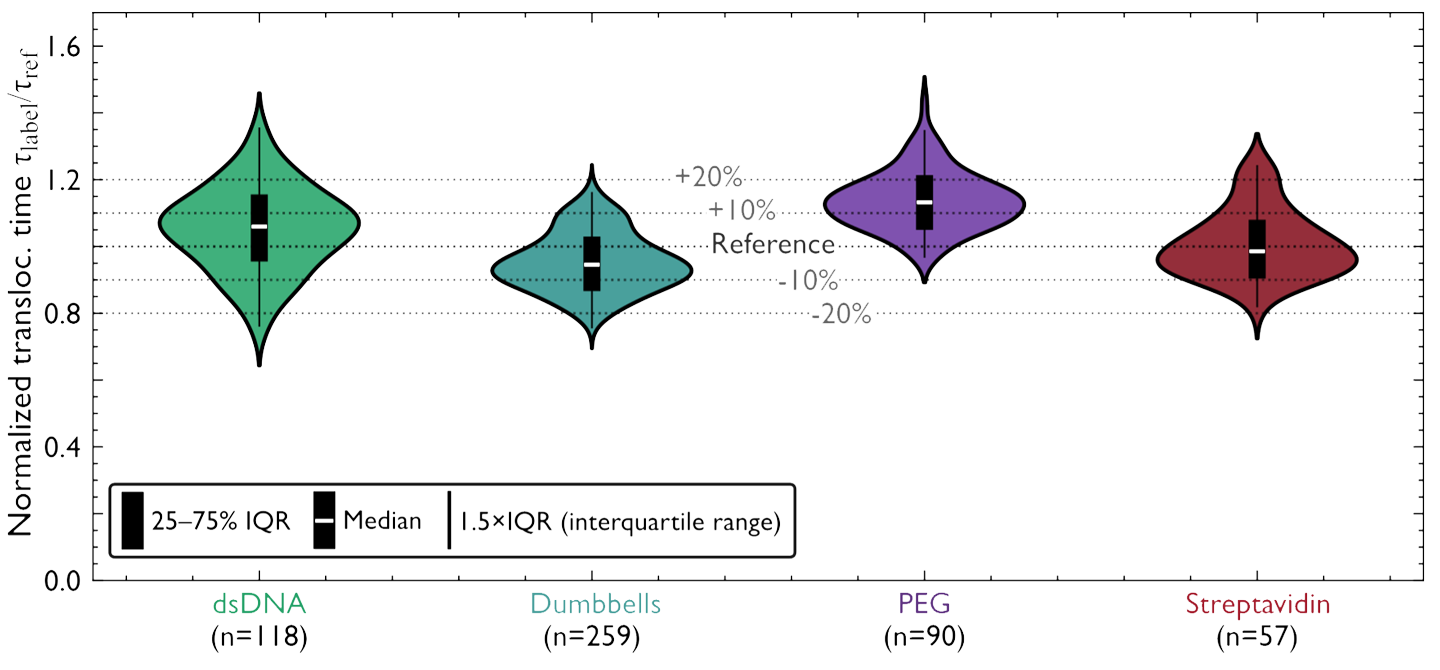
Before the start of the DYNAMO project, nanopore technologies and DNA nanotechnology had already demonstrated strong potential for single-molecule sensing and sequencing applications[cite: 14]. Solid-state nanopores were widely investigated because of their stability, tunable geometry, and compatibility with electrical detection techniques [1,2][cite: 14]. At the same time, DNA origami emerged as a powerful platform for constructing programmable nanoscale architectures with controllable geometries and functionalities [3,4][cite: 14]. Early studies demonstrated that DNA-origami nanopores could be integrated with solid-state membranes to create hybrid systems with improved sensitivity and controllable molecular transport [5,6][cite: 14]. Bell et al. (2012) first showed that DNA-origami structures could be docked into and ejected from solid-state nanopores under applied voltage, enabling the resistive-pulse detection of λ-DNA through hybrid pores [5][cite: 14]. Subsequent work extended the concept to graphene membranes and to programmable DNA nanoswitches read out by ionic current [6,7][cite: 14]. However, recurring limitations were reported, including residual leakage currents through the DNA scaffold, elevated and variable ionic noise compared with bare solid-state pores, and only partial control over the orientation and stability of the inserted origami [2][cite: 14].
Electroosmotic flow in nanopores and nanocapillaries
Electroosmotic flow (EOF) had also already been identified as an important mechanism governing transport inside nanopores and nanocapillaries [8,9][cite: 14]. Previous studies showed that EOF could influence translocation dynamics, trapping efficiency, and ionic transport near charged surfaces [9,10][cite: 14]. Asandei et al. (2016) demonstrated that EOF could be used as an electroosmotic trap against the electrophoretic force to capture peptides at a protein nanopore [10][cite: 14], and similar trapping concepts were extended to whole proteins inside engineered biological pores [11][cite: 14]. In glass nanocapillaries, Laohakunakorn and Keyser established that EOF profiles depend strongly on pore geometry and ionic strength, with flow reversal observed outside conical pores when the salt concentration is lowered [9,12][cite: 14]. However, most experimental investigations were performed under relatively limited conditions, often focusing on a single salt type (typically KCl), narrow concentration ranges, or comparatively large capillary geometries with limited spatial resolution of the flow field[cite: 14]. As a result, the detailed dependence of EOF on ion identity, concentration, and nanoscale confinement remained insufficiently understood, and three-dimensional mapping of the flow field outside a nanopore had only just been demonstrated as a proof of concept [13][cite: 14].
Thermally responsive DNA nanostructures and hybridization physics
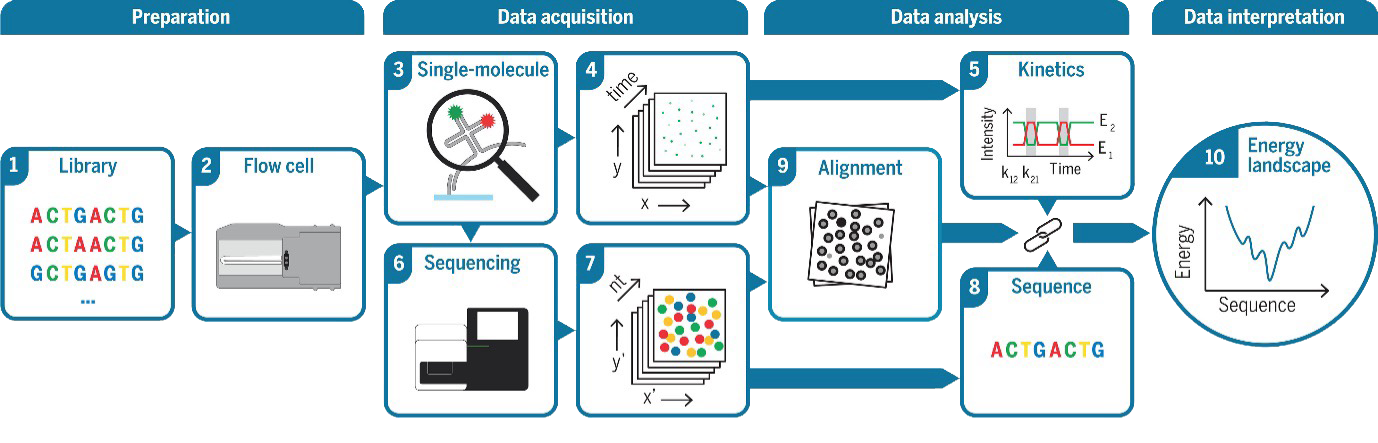
Thermally responsive DNA nanostructures and reversible DNA melting had already demonstrated the possibility of controllable structural switching at the nanoscale [14,15][cite: 14]. DNA origami nanocapsules and lattices reconfigurable by pH, temperature, or strand displacement had been reported, often relying on toehold-mediated strand exchange or triplex-forming pH latches [14,15,16][cite: 14]. Nevertheless, most earlier studies primarily focused on proof-of-concept demonstrations of structural reconfiguration, while the underlying physics of DNA hybridization remained only partially understood, especially under confined nanopore conditions and at the single-molecule level[cite: 14]. Theoretical and coarse-grained simulation work had outlined a picture of hybridization proceeding through non-specific contact, stochastic nucleation, and zipping [17,18][cite: 14], but direct experimental access to these elementary steps for short, surface-tethered strands embedded in nanofluidic environments was still largely missing[cite: 14].
References
- [1] Dekker, C. Solid-state nanopores. Nat. Nanotechnol. 2, 209–215 (2007)[cite: 14].
- [2] Hernández-Ainsa, S. & Keyser, U. F. DNA-origami nanopores: developments, challenges and perspectives. Nanoscale 6, 14121–14132 (2014)[cite: 14].
- [3] Rothemund, P. W. K. Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006)[cite: 14].
- [4] Dietz, H., Douglas, S. M. & Shih, W. M. Folding DNA into twisted and curved nanoscale shapes. Science 325, 725–730 (2009)[cite: 14].
- [5] Bell, N. A. W. et al. DNA origami nanopores. Nano Lett. 12, 512–517 (2012)[cite: 14].
- [6] Barati Farimani, A. et al. DNA origami–graphene hybrid nanopore for DNA detection. ACS Appl. Mater. Interfaces 9, 92–100 (2017)[cite: 14].
- [7] Bell, N. A. W. & Keyser, U. F. Specific protein detection using designed DNA carriers and nanopores. J. Am. Chem. Soc. 137, 2035–2041 (2015)[cite: 14].
- [8] Firnkes, M. et al. Electrically facilitated translocations of proteins through silicon nitride nanopores: conjoint and competitive action of diffusion, electrophoresis, and electroosmosis. Nano Lett. 10, 2162–2167 (2010)[cite: 14].
- [9] Laohakunakorn, N. et al. Electroosmotic flow reversal outside glass nanopores. Nano Lett. 15, 695–702 (2015)[cite: 14].
- [10] Asandei, A. et al. Electroosmotic trap against the electrophoretic force near a protein nanopore reveals peptide dynamics during capture and translocation. ACS Appl. Mater. Interfaces 8, 13166–13179 (2016)[cite: 14].
- [11] Schmid, S. et al. Nanopore electro-osmotic trap for the label-free study of single proteins and their conformations. Nat. Nanotechnol. 16, 1244–1250 (2021)[cite: 14].
- [12] Mc Hugh, J., Andresen, K. & Keyser, U. F. Cation-dependent electroosmotic flow in glass nanopores. Appl. Phys. Lett. 115, 113702 (2019)[cite: 14].
- [13] Mc Hugh, J., Thorneywork, A. L., Andresen, K. & Keyser, U. F. 3D flow field measurements outside nanopores. Rev. Sci. Instrum. 93, 053703 (2022)[cite: 14].
- [14] Ijäs, H. et al. Reconfigurable DNA origami nanocapsule for pH-controlled encapsulation and display of cargo. ACS Nano 13, 5959–5967 (2019)[cite: 14].
- [15] Kosuri, P., Altheimer, B. D., Dai, M., Yin, P. & Zhuang, X. Rotation tracking of genome-processing enzymes using DNA origami rotors. Nature 572, 136–140 (2019)[cite: 14].
- [16] Marras, A. E., Zhou, L., Su, H.-J. & Castro, C. E. Programmable motion of DNA origami mechanisms. Proc. Natl. Acad. Sci. USA 112, 713–718 (2015)[cite: 14].
- [17] Ouldridge, T. E. et al. DNA hybridization kinetics: zippering, internal displacement and sequence dependence. Nucleic Acids Res. 41, 8886–8895 (2013)[cite: 14].
- [18] SantaLucia, J. & Hicks, D. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 33, 415–440 (2004)[cite: 14].
- Hits: 209