Project 6
- ESR Student Name: Hiba Haneena Karakkal
- Hits: 3
Proteins govern life. They carry oxygen, catalyse reactions, transmit signals, and form the structural fabric of every cell. Understanding how individual proteins behave, how they fold, interact, and function is fundamental to medicine, diagnostics, and drug discovery. Yet most tools available today require attaching a synthetic fluorescent dye to a protein before it can be detected. This labelling step, while effective, is not neutral: it can alter the protein's shape, disrupt its binding interactions, and distort the very behaviour it was designed to reveal.
Single-molecule fluorescence techniques like FCS, FRET, FLIM, and STED have transformed our understanding of molecular biology. However, they almost universally depend on extrinsic fluorescent labels. The field has long sought an alternative rooted in a simple observation: proteins are not dark. Aromatic amino acids like tryptophan, tyrosine, and phenylalanine emit an autofluorescence under ultraviolet (UV) illumination. More than 90% of all proteins contain at least one such residue.
Exploiting this intrinsic UV autofluorescence for single-molecule detection has been a long-standing challenge. Proteins are orders of magnitude dimmer than conventional dyes, and UV microscopy introduces specific difficulties: rapid photobleaching, high background from buffer and optics, and the absence of nanophotonic platforms optimised for the UV spectral range. Early efforts were limited to large proteins carrying dozens of tryptophan residues, a restriction incompatible with most biologically relevant targets.
Recent years have seen decisive progress. The Wenger group at Institut Fresnel demonstrated that UV-optimised plasmonic nanoantennas, including aluminium nanoapertures, optical horn antennas, and rhodium nanocube dimers, can enhance UV autofluorescence sufficiently to resolve single proteins containing as few as one tryptophan residue (Roy et al., Nano Letters 2023; Barulin et al., Nature Communications 2022). Zero-mode waveguides (ZMWs), nanoscale apertures that confine light below the diffraction limit, have also been shown to boost brightness and enable operation at physiologically relevant micromolar concentrations (Barulin et al., Nano Letters 2019). In parallel, plasmonic nanopores have emerged as powerful single-molecule sensors capable of monitoring the translocation of individual biomolecules with optical readout, moving beyond the limitations of conventional ionic current detection (Verschueren et al., ACS Nano 2019; Zhao et al., ACS Photonics 2022).
Despite these advances, combining UV autofluorescence with nanophotonic enhancement and nanopore-based translocation in a unified, label-free platform remains an open frontier and is precisely what this project addresses.
My project under DYNAMO is centred on UV autofluorescence correlation spectroscopy (UV-FCS), a technique that measures the natural UV emission of individual proteins as they diffuse through a tiny illuminated volume, revealing information about their concentration, size, and dynamics. The intrinsic signal is weak, so two complementary strategies are deployed: reducing noise and amplifying the signal.
First is suppressing the background with FLCS. Fluorescence Lifetime Correlation Spectroscopy (FLCS) adds a time dimension to the measurement and can mathematically separate genuine signal from scattered laser light and other parasitic contributions. This lifetime-gating approach substantially improves the signal-to-background ratio without any modification to the protein or sample.
The second strategy is amplifying the signal with nanophotonics. Zero-mode waveguides (ZMWs) are nanoscale apertures fabricated in metal films confining light to volumes below the diffraction limit. When a protein enters this ultrasmall hotspot, its fluorescence is dramatically enhanced. This makes it possible to detect individual proteins that would otherwise be invisible, and to work at the physiologically relevant concentrations found in living systems.
Further, we plan to extend the detection platform to plasmonic nanopores: nanoscale channels that force proteins to pass through one at a time, driven by an electric field, and give electrical readouts. But our focus is on the optical readout strategy. Each translocation event produces a distinct optical burst, carrying information not only about the protein's presence but also about its dynamics and identity. Combined with the UV autofluorescence readout, this approach opens the possibility of characterising proteins with single-molecule resolution, label-free, in real time.
Together, these tools form a coherent label-free platform for single-protein analysis. The integration of UV autofluorescence with nanopore translocation could enable a new class of biosensor, one capable of identifying proteins by their intrinsic optical fingerprint as they thread through a nanoaperture. This would have direct implications for medical diagnostics, proteomics, and the study of protein misfolding diseases such as Alzheimer's and Parkinson's.
Solid-state nanopore devices are potential platforms for single-molecule detection and sequencing. These devices detect entities such as molecules, nanoparticles, or nanoparticles tagged with molecules as they travel across a thin membrane via nanopore. Their enticement originates from the ability to investigate entities using extremely small sample volumes and without the need for extensive preparation. However, conventional nanopore techniques had a significant limitation- molecules often traversed the pore very quickly and in a stochastic fashion, providing only a little time window for study. This rapid translocation made it challenging to obtain robust and consistent signals, particularly for advanced applications like molecular identification, extended optical measurements, and sequencing.
Plasmonic nanopores significantly improved conventional solid state nanopore devices by integrating metallic nanostructures. These metallic structures can confine light, allowing optical detection methods like as fluorescence and Raman spectroscopy. This enabled the combination of electrical and optical readouts, which improved sensitivity significantly. However, plasmonic nanopores presented new obstacles. Strong optical fields can generate local heating, disrupt the liquid environment and prevent steady trapping or measurement. Furthermore, optical or plasmonic forces alone are insufficient to ensure stable, regulated and robust entrapment of tiny particles or tagged biomolecules near the pores.
Our group earlier proposed a theoretical concept, a hybrid magnetoplasmonic nanopore design with a thin cobalt layer sandwiched between two gold layers. The goal was to combine plasmonic optical enhancement and magnetic control in a single nanopore platform. In this notion, an external magnetic field might configure the magnetic layer, resulting in localized magnetic tweezers near the nanopore edge. These magnetic tweezers were expected to catch magnetic core-shell nanoparticles near to the pore wall, extending observation time and aiding particle positioning. This work anticipated magnetic trapping forces of up to 28 pN for a 10 nm magnetite nanoparticle, which was substantially higher than the forces generally obtained using standard optical or plasmonic trapping methods.
This theoretical work explained that magnetism could overcome one of the fundamental limitations of nanopores: a lack of active control over analyte location and movement. It also proposed that placing a magnetoplasmonic nanoparticle close to the nanopore wall could result in a small nanocavity with improved optical fields. However, at that point, the technique remained theoretical. The optical enhancement inside the original nanopore design was still restricted, and the integration of cobalt resulted in higher optical losses when compared to a pure gold nanopore. As a result, the field had a compelling concept, but no empirically validated platform that integrated plasmonic enhancements, magnetic trapping in a single device. The main open challenges were to experimentally realize such hybrid devices, improve the optical signal, reduce limitations associated with local heating or weak trapping, and design architectures capable of plasmonic enhancements and controlling particles at the same nanopore which we explored in this work.
Naveen Kumar (JR3) | University of Leipzig | Supervisor: Prof. Ralf Seidel
By 2022, DNA origami had matured into a versatile three-dimensional construction platform with sub-nanometre positional precision[cite: 41]. At Leipzig, Seidel and co-workers had pioneered DNA origami molds for shape-controlled metallic nanoparticle synthesis — casting gold nanorods inside tubular cavities (Helmi et al., 2014) and developing a modular construction kit for complex metal nanostructures[cite: 42]. The platform had been extended to palladium nanostructures (Ye et al., 2021)[cite: 43]. DNA origami nanopores had been demonstrated in solid-state platforms and lipid bilayers[cite: 44]. However, the integration of thermal control with these nanostructures — to reversibly gate molecular access or dynamically reconfigure sensing elements — remained unrealised[cite: 45].
Equilibrium thermodynamics of DNA duplex formation were well described by the nearest-neighbour model (SantaLucia, 1998), but kinetic parameters — association and dissociation rates — were poorly characterised[cite: 47]. Single-molecule techniques (smFRET, DNA-PAINT) provided dwell-time information for specific systems, but systematic temperature-dependent measurements across diverse sequences were absent[cite: 48]. Whether association rates increased, decreased, or remained constant with temperature was actively debated[cite: 49]. No high-throughput platform existed to measure sequence-specific kon and koff across temperatures simultaneously[cite: 50].
Toehold-mediated strand displacement (Zhang & Winfree, 2009) had become the standard mechanism for dynamic DNA nanotechnology[cite: 52]. While displacement kinetics had been characterised in bulk at room temperature (Srinivas et al., 2013; Irmisch et al., 2020), the temperature dependence of displacement rate constants and the activation barriers governing strand invasion and branch migration were not systematically characterised[cite: 53]. This gap was critical for designing thermally switchable DNA devices that must operate across a range of temperatures[cite: 54].
Surface-enhanced Raman scattering (SERS) in plasmonic nanostructures had achieved single-molecule sensitivity[cite: 56]. DNA origami provided scaffolds for assembling nanoparticle dimers with defined gap sizes (~2–10 nm), creating electromagnetic hotspots with enhancement factors exceeding 108. However, systematic methods for reproducible analyte positioning and orientation control within hotspots were lacking[cite: 57]. The DNA origami mould technology offered a promising route for casting metal nanostructures with programmable gap geometries, but quantitative integration with thermally switchable DNA elements had not been demonstrated[cite: 58].
Fluorescence thermometry using Rhodamine B and other temperature-sensitive dyes had been demonstrated in microfluidic contexts, but integration with TIRF microscopy for surface-temperature calibration during single-molecule experiments was not established[cite: 60]. Sub-diffraction temperature mapping — essential for characterising thermal gradients around plasmonic heaters — remained an unmet challenge[cite: 61].
Before the DYNAMO project began, researchers could already make simple solid-state nanopores by drilling holes in thin silicon nitride membranes using a focused ion beam or electron beam lithography. Typical pore diameters were around 10–30 nm, and the membranes were about 20–100 nm thick. However, making many identical nanopores in an array was still difficult because each pore had a slightly different size and shape. This lack of reproducibility was a major bottleneck.
Some efforts had been made to use plasmonic structures for surface‑enhanced Raman spectroscopy (SERS) in nanopores. By placing metal films or nanoparticles around the pore, researchers could obtain stronger Raman signals from molecules passing through. However, the acquisition frequency of Raman spectra was typically very low – often on the order of seconds or even longer per spectrum. This made it impossible to capture fast, transient events such as a single molecule translocating through the pore in microseconds. In addition, almost all of these works used the plasmonic structure passively, only to boost the signal. There was very little exploration of using the plasmonic structure for active control, for example by shining a laser to generate local heating (plasmonic heating) and deliberately switching the pore’s behavior. The idea of using light not just to see but also to manipulate what happens inside the nanopore remained largely unexplored.
Mixing different functional materials with nanopores was still at an early stage. Magnetic materials (such as cobalt or nickel) and plasmonic metals had been combined in theory, but very few experimental devices existed. People had started to use two-dimensional materials like graphene or MoS₂ as the membrane itself, but applications of hybrid channels where a 2D material sits on top of a silicon nitride trench were not widely explored. DNA origami – folded DNA that can position metal nanoparticles with nanometer precision – had been demonstrated in solution, but attaching it reliably to a solid-state nanopore and keeping it stable during electrical measurements remained a challenge.
When it came to controlling the movement of ions or molecules through a nanopore, most approaches were passive. They relied on fixed properties such as the pore size, the surface charge, or the salt concentration and pH of the buffer solution. These methods worked for steady‑state measurements, but they could not actively switch the pore on and off on demand. The use of external stimuli to achieve active control – such as fast optical heating, real‑time voltage‑driven chemical reactions, or gate‑voltage tuning of surface charge – had not yet been widely explored. In general, the field lacked fast, reversible, and localized active gating mechanisms.
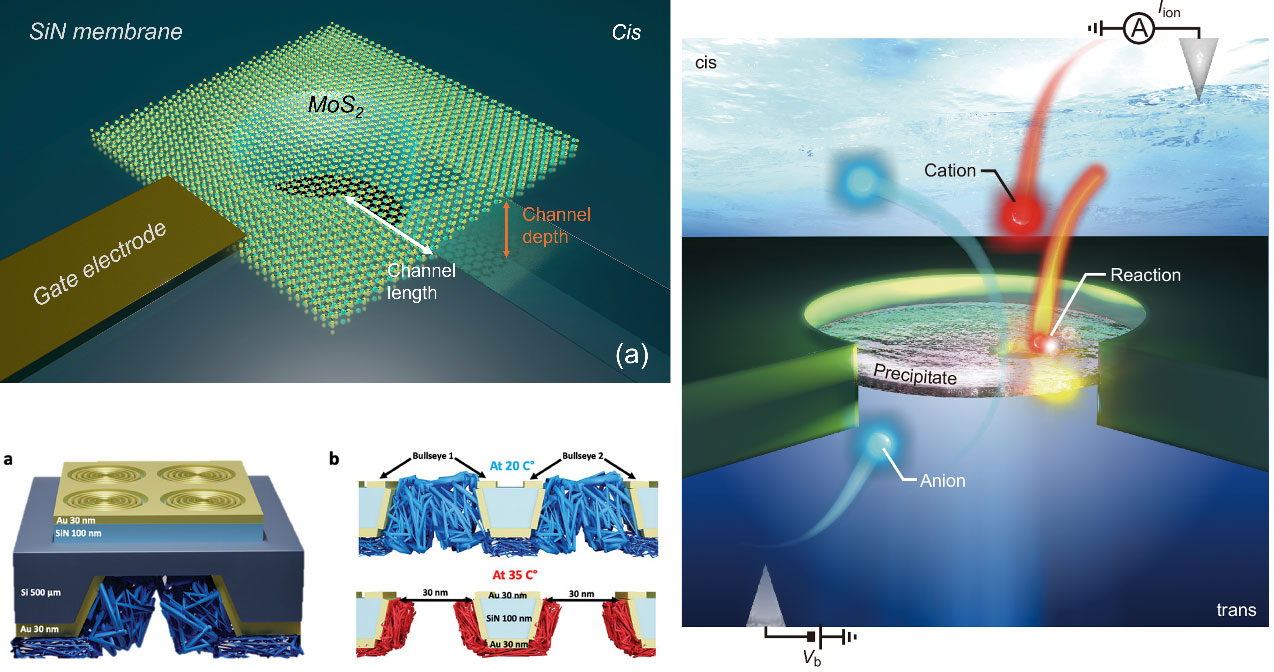
As a Doctoral Researcher in the DYNAMO network, Shukun Weng extended his expertise from nanostructure textured perovskite devices to hybrid plasmonic nanopores and nanofluidic devices. He contributed to the design and fabrication of several gating platforms that control ion transport. In one work, he integrated a thermoresponsive polymer (PNIPAM) with a gold bullseye plasmonic structure. Laser illumination creates a local temperature control, triggering reversible polymer swelling/collapse that opens or closes the nanopore within milliseconds. This optothermal gate achieves an on/off ratio of 60 and enables selective addressing of individual pores in an array, demonstrating logic operations.
In another line, he helped develop voltage-controlled in-pore chemistry: electromigration of Ca²⁺ or Mn²⁺ from the cis side into a nanopore containing phosphate buffer induces reversible precipitation/dissolution of metal phosphates. This creates a nanofluidic diode with a rectification ratio exceeding 40,000 and a memristor operating at sub-nanowatt power. Using a plasmonic bullseye nanopore, he performed surface-enhanced Raman spectroscopy (SERS) to directly monitor the precipitate formation inside the attoliter-scale pore – the first real-time chemical fingerprinting of such dynamic reactions.
He also developed a MoS₂/SiN hybrid nanochannel (≈10 nm height, 100 nm width) where a gate voltage tunes the surface charge of MoS₂, enabling ambipolar ion transport. This device harvests osmotic power from a salt gradient, achieving a power density of 18 kW/m² from a single channel, and detects unfolded BSA proteins with significantly extended dwell times due to MoS₂-protein interactions. Collectively, these contributions establish a family of electrically, optically, and chemically gated nanopores that are scalable, energy-efficient, and suitable for future iontronic circuits.